L’étude de plus en plus aisée des écosystèmes microbiens – on parle de microbiotes – offre de nouvelles perspectives d’applications, dans des domaines aussi variés que l’agriculture ou la santé humaine ou encore la lutte contre le bioterrorisme, la détection d’agents pathogènes émergents, et la caractérisation de leurs réservoirs.
La
métaprotéomique permet d’identifier rapidement le contenu protéique d’un microbiote et de sonder comment il fonctionne. Théoriquement, il est également possible d’établir la diversité des microorganismes[1] qui le composent. Les branches de la vie et niveaux taxonomiques (souche, espèce, genre, famille…) des peptides identifiés par spectrométrie de masse peuvent être déterminés en comparant leurs séquences à celles contenues dans des bases de données moléculaires. Mais il existe des limites. La robustesse et l’exhaustivité de la base de données utilisée influencent directement le résultat, la plupart des peptides étant partagés entre microorganismes. Notamment, plus les bases de données s’enrichissent, plus il devient ardu d’y retrouver ses petits.
Une équipe du Laboratoire Innovations technologiques pour la Détection et le Diagnostic (LI2D, à Marcoule) du Département Médicaments et Technologies pour la Santé (DMTS) vient de développer une méthode mathématique pour identifier les organismes présents dans un microbiote et les doser. Elle consiste à prédire pour chaque organisme vivant le nombre de peptides communs à tous les organismes présents dans la base de données et de considérer que l’ensemble du signal obtenu par spectrométrie de masse pour un microbiote est en fait une combinaison des signatures de tous les organismes qui le composent.
Les chercheurs ont analysé le protéome de l’espèce
Shigella flexneri par spectrométrie de masse (tandem MS/MS) et ont comparé les séquences peptidiques à une base de données. Ils ont alors déterminé le nombre de peptides de la base de données assignés à
Shigella flexneri qu’ils retrouvaient effectivement dans les données de spectrométrie, ce qu’ils appellent le nombre de correspondances entre le taxon et le spectre (TSM,
taxon-to-spectrum matches). Puis, ils ont refait la même comparaison, en considérant non pas les peptides de la base assignés à
Shigella flexneri mais ceux attribués à d’autres espèces, plus ou moins éloignées phylogénétiquement de
Shigella flexneri. Ainsi, une espèce très proche de
S. flexneri (comme
Escherichia coli) a globalement plus de peptides communs avec elle, et donc un nombre de TSM plus élevé, qu’une espèce très éloignée (comme
Bacillus subtilis). En comparant toutes les données, les chercheurs ont mis en évidence une corrélation entre le nombre de TSM et la distance phylogénétique entre les espèces. Pour valider la méthode et montrer qu’elle est quantitative, les chercheurs l’ont ensuite éprouvée en analysant d’abord des mélanges artificiels de deux organismes proches phylogénétiquement,
S. flexneri et
Salmonella bongori, avec des proportions différentes des deux, puis des modèles de microbiotes plus complexes.
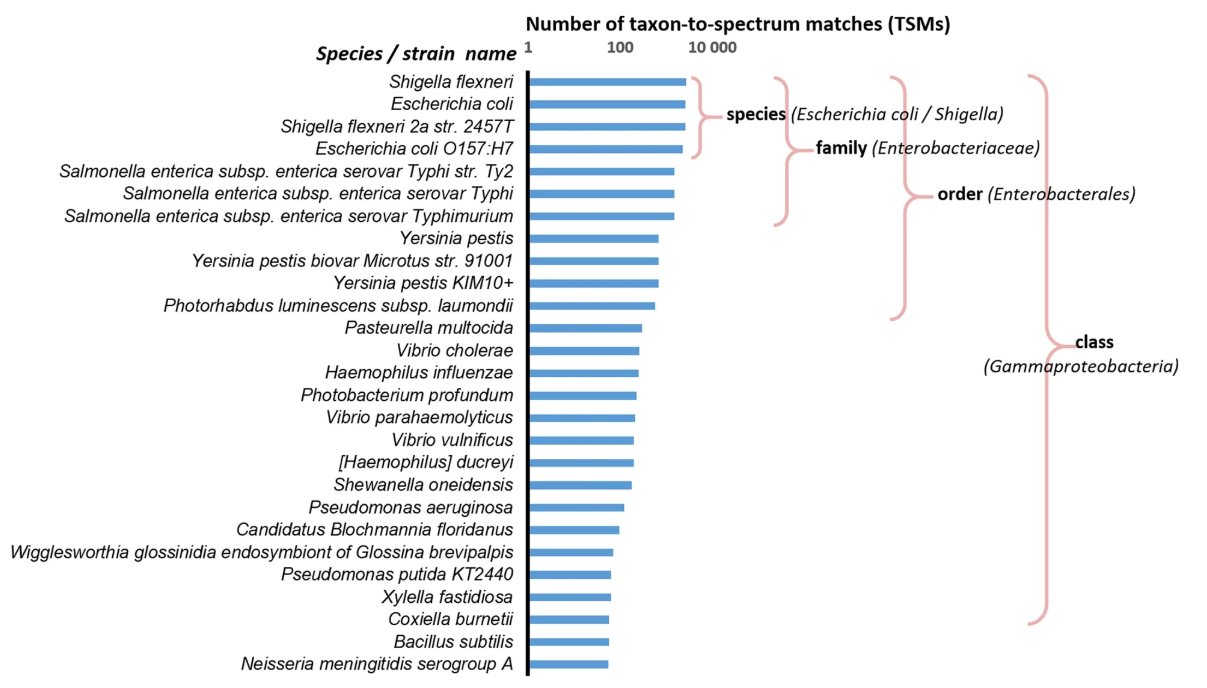
Nombre de correspondances entre les taxons et le spectre (TSM) pour un échantillion pur de
Shigella flexneri. Les données du spectre qui peuvent être associées aux peptides et protéines d'une sélection de taxons disponibles dans une base de donnée (Ciccarelli et al. ) sont dénombrés. Une observation clé est que le nombre de TSM attribués à des taxons qui ne font pas partie de l'échantillon est directement lié à la proximité taxonomique vis à vis de Shigella flexneri. O. Pible et al. Microbiome, 2020
Cette nouvelle méthode d’analyse, qu’ils nomment phylopeptidomique, ouvre de nouveaux horizons pour l’étude des microbiotes et de leur dynamique. La méthode peut s’appliquer aussi bien pour les bactéries, que les archées, ou les eucaryotes. Elle permet une identification sans a priori et rapide de tout organisme vivant, et ce même en mélange complexe. Dans le cadre du programme interministériel de recherche et développement contre le risque terroriste NRBC-E (Nucléaire, Radiologique, Biologique, Chimique et Explosif), le laboratoire développe des applications de cette nouvelle méthode à l’identification sans a priori de tous types d’agents pathogènes.
Contact :
Jean Armengaud
[1] La majorité des protéines sont conservées au sein d’un genre microbien, mais d’une espèce à l’autre, d’une souche à l’autre, des variations de leur séquence d’acides aminés existent.