Elucider, prédire et inhiber les interactions protéine-protéine impliquées dans le maintien de l'intégrité du génome
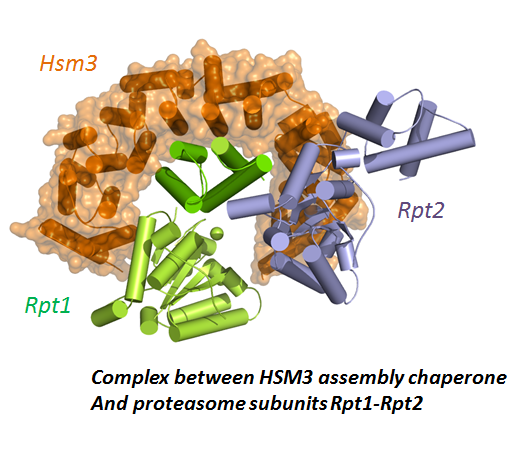
Les complexes protéiques sont au cœur de la plupart des processus biologiques. Notre projet vise à découvrir comment ces interactions physiques assurent les régulations croisées entre multiples machineries et voies de signalisation dans les réponses des cellules aux stress génotoxiques. Notre équipe s’intéresse particulièrement aux machineries protéiques modulant l’établissement de l’information épigénétique et à celles impliquées dans les processus de recombinaison. Nous combinons l’utilisation de techniques expérimentales comme la RMN et cristallographie des rayons X avec le développement d’approches bioinformatiques originales pour le docking moléculaire qui augmentent la qualité des modèles structuraux de complexes. L’inhibition de ces interactions par des composés exogènes émerge comme un champ de recherche particulièrement excitant pour le développement de nouvelles stratégies thérapeutiques. Dans cette optique, nous entreprenons le design de composés qui inhibent la formation des assemblages protéiques activés suite à des stress génotoxiques.
Caractérisation structurale des complexes en combinant RMN, cristallographie et modélisation
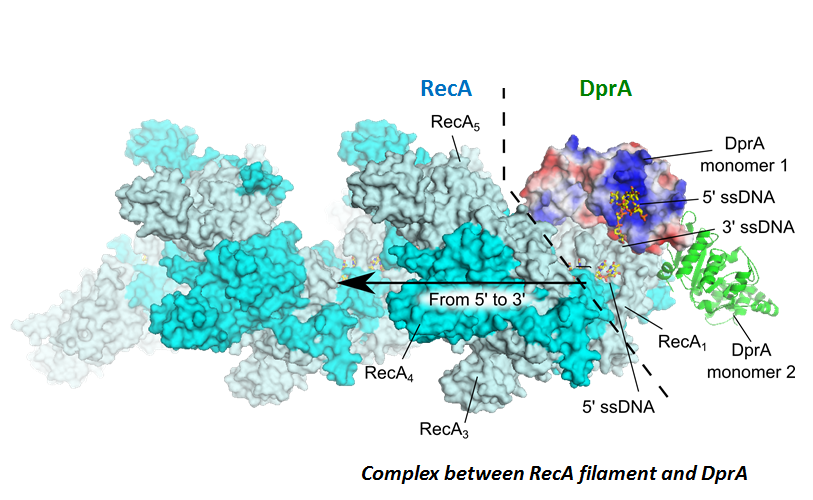
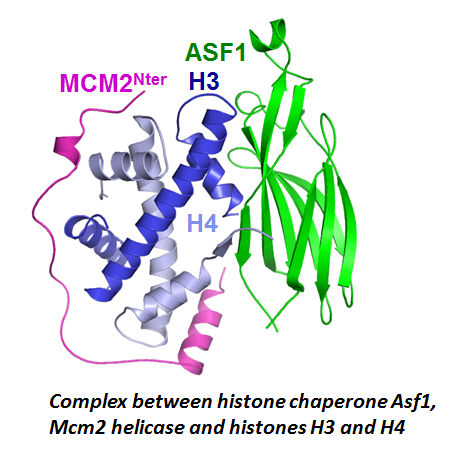 Le maintien de l’intégrité du génome repose sur un ensemble de voies de signalisation et de machineries qui communiquent entre elles par de multiples interactions physiques. Comprendre la logique moléculaire associée à ces interactions nécessite de caractériser les synergies et compétitions activées en réponse à des stress cellulaires tels que l’exposition à des agents génotoxiques. Dans ce cadre, nous nous intéressons à une classe de protéines particulièrement importante pour l’assemblage des machineries et des complexes macromoléculaires, les chaperons d’assemblages. Nous avons par exemple montré comment Asf1, un chaperon central des histones H3 et H4 est capable de s’associer de façon synergique avec les histones et l’hélicase réplicative Mcm2 pour coordonner la manipulation des composants du nucléosome au plus près de la fourche de réplication. D’autres surfaces constituent des sites sur lesquels des partenaires comme Rad53 (signalisation des dommages de l’ADN), HirA (transcription) ou CAF1 (réplication) interagissent de façon compétitive. L’ensemble de ces interactions est activé dans différents contextes épigénétiques. Nous cherchons désormais à comprendre comment s’effectue le couplage entre les reconnaissances moléculaires de ces contextes épigénétiques et l’activation de réponses cellulaires spécifiques.
Le maintien de l’intégrité du génome repose sur un ensemble de voies de signalisation et de machineries qui communiquent entre elles par de multiples interactions physiques. Comprendre la logique moléculaire associée à ces interactions nécessite de caractériser les synergies et compétitions activées en réponse à des stress cellulaires tels que l’exposition à des agents génotoxiques. Dans ce cadre, nous nous intéressons à une classe de protéines particulièrement importante pour l’assemblage des machineries et des complexes macromoléculaires, les chaperons d’assemblages. Nous avons par exemple montré comment Asf1, un chaperon central des histones H3 et H4 est capable de s’associer de façon synergique avec les histones et l’hélicase réplicative Mcm2 pour coordonner la manipulation des composants du nucléosome au plus près de la fourche de réplication. D’autres surfaces constituent des sites sur lesquels des partenaires comme Rad53 (signalisation des dommages de l’ADN), HirA (transcription) ou CAF1 (réplication) interagissent de façon compétitive. L’ensemble de ces interactions est activé dans différents contextes épigénétiques. Nous cherchons désormais à comprendre comment s’effectue le couplage entre les reconnaissances moléculaires de ces contextes épigénétiques et l’activation de réponses cellulaires spécifiques.
Nouveaux outils de prédiction de structure des complexes par l’intégration des contraintes évolutives

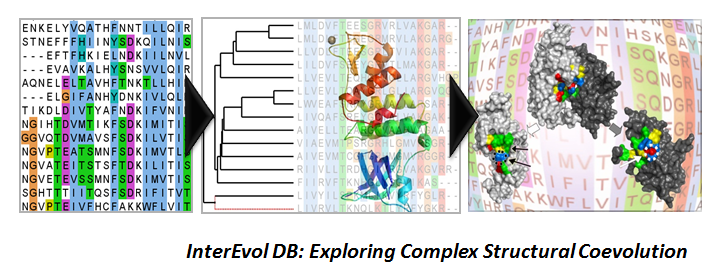
Au cours des dernières années, notre équipe a significativement amélioré les méthodes de prédiction de structures des complexes protéiques en intégrant la dimension évolutive aux outils traditionnels de docking moléculaire. Grâce au développement et à l’exploitation de la base de données InterEvol, nous pouvons explorer les propriétés évolutives des interfaces de complexes protéiques à l’échelle structurale. L’analyse de plus de 1000 couples de complexes dits interologues (impliquant des sous-unités homologues entre elles) a révélé les propriétés des interfaces les plus conservés au cours de l’évolution et les conséquences structurales des processus de co-évolution. Le développement du score de docking InterEvScore, combinant un potentiel statistique à deux et trois corps avec l’information de co-évolution, permet de renforcer la fiabilité de nos prédictions. Ces développements nous ont permis d’obtenir de très bons résultats pour plusieurs cibles proposées dans l’expérience CAPRI (Critical Assessment of Prediction of Interactions) en particulier lors de la
conférence CASP11.
Nos études actuelles visent à comprendre comment la richesse d’information contenue dans les alignements multiples de séquences peut être exploitée par de nouvelles approches afin d’augmenter la qualité des modèles structuraux de complexes protéiques.
Design de nouveaux composés inhibiteurs des interactions protéine-protéine
L’inhibition des interactions protéine-protéine constitue un défi méthodologique qui repose sur des innovations importantes en modélisation, chimie biomimétique, biologie structurale et caractérisation cellulaire. Nous avons investi ce champ de recherche dans l’optique d’inhiber l’action des chaperons d’histones comme Asf1 qui constitue une cible anti-cancéreuse nouvelle et prometteuse. Nous avons déjà développé un peptide de première génération capable d’inhiber l'interaction entre Asf1 et les histones. Ce peptide pénètre efficacement dans les cellules où il empêche la prolifération en bloquant la progression du cycle cellulaire et induit la mort de lignées tumorales en culture. Basé sur ces résultats préliminaires particulièrement encourageants, nous concevons des composés peptido-mimétiques, plus puissants et plus sélectifs, capable d’inhiber l'activité d’Asf1 et caractérisons leur potentiel thérapeutique.