Our group conducts research in the field of computational biology and chemistry. In particular, we apply theoretical techniques (such as classical and quantum Molecular dynamics (MD) simulations) to develop models and software for studying challenging biophysical phenomena closely related to biophysics and biology.
Team Leader
Massimo MARCHI
massimo.marchi[at]cea.fr
We pursue the following research themes:
- Development of force fields for MD simulations of protein-detergent complexes,
- Molecular modelling and simulations of membrane photosynthetic complex,
- Structure and dynamics of the hydration around proteins or surfactant assemblies.
Development of force fields for MD simulations of protein-detergent complexes
To investigate the structural properties of peptides/proteins membrane in solution, it is necessary to use accurate amphiphilic molecules (e.g. detergents). In this context, we are developing innovative approaches to parameterize molecular potential (i.e. force fields) for MD simulations. For example, by using an approach called "molecular building blocks" (Fig. 1, left) where a surfactant molecule is cut in different molecular fragments with a well-defined conformations. With this approach, we recently developed accurate models for glycolipid-based (coll. Pr. Alex Mackerell, University of Maryland School of Pharmacy, USA) or n-alkyl phosphocholine (Coll. Pr. François-Yves Dupradeau, (LG2A) FRE 3517 CNRS, University of Amiens, France) surfactants compatible with the AMBER force fields for proteins. These force fields have been validated by characterizing with extensive MD simulations the structural properties of pure micelle dodecyl-maltoside (DDM) (Fig. 1, right) and dodecyl phosphocholine (DPC) in the presence or membrane peptides from a ABC-type membrane protein (MRP1/ABCC1) involved in cellular detoxification (Coll. Dr. B. de Foresta, LPM, CEA) (Fig. 2)4.
We are pursuing this research area on developing a large detergent library in order to examine the influence of the surfactant environment on peptide/protein structure models.
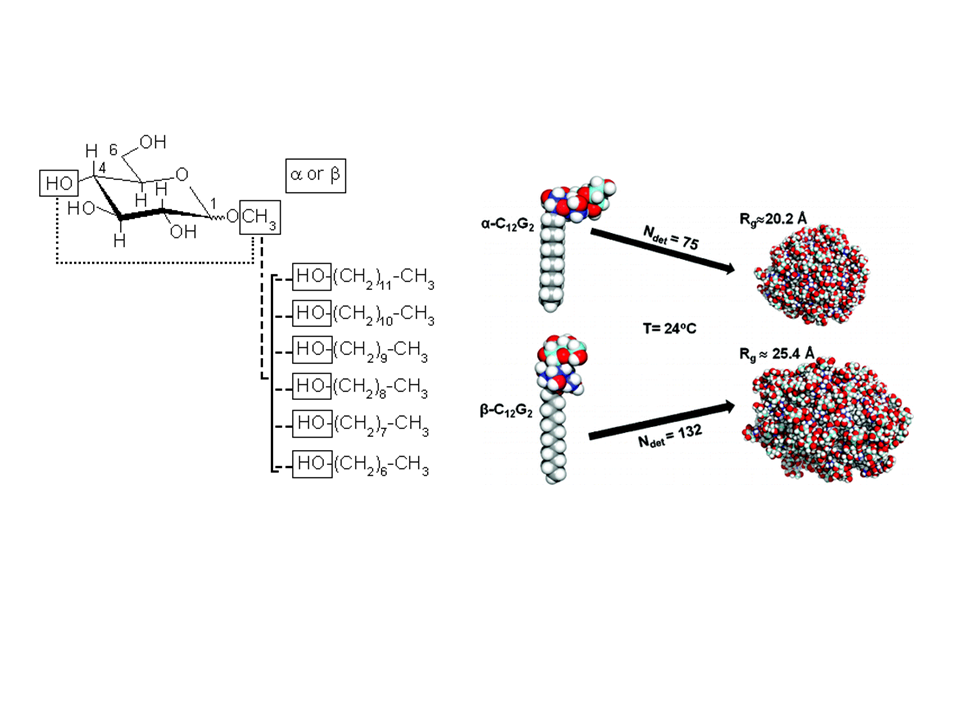
Fig. 1: (Left) Example of molecular building block approach applied to derive atomic partial charges for glycolipid-based surfactants. DDM micelle structures as a function of the surfactant headgroup conformation ©CEA/S.Abel.
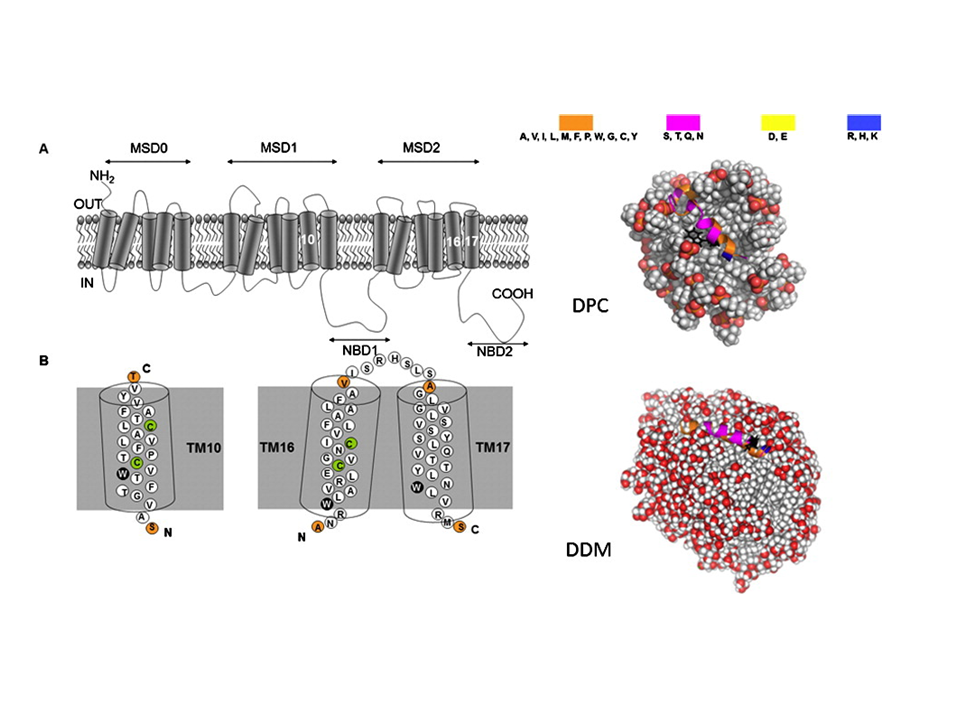
Fig. 2 (Left) Topology of the hMRP1 membrane protein showing its transmembrane domains and examples of TM10 fragment binding to the DPC and DDM micelle models (right). ©CEA/S.Abel.
We also collaborate with different national and international labs that use MD simulations for studying bolaform surfactants assemblies (coll. Dr. Niki Baccile, (LCMCP), Collège de France, Paris, France) or the solubilization of polycyclic aromatic compounds into micelles (coll. Xujun Liang, South China University of Technology, China).
Simulations of membrane photosynthetic complex
An important part of our current research activity also concerns the elucidation by theoretical means of the molecular mechanism of electron transfer (ET) in bacterial photosynthetic membrane complexes. To this end, we have developed force fields for the photosynthetic cofactors (such as chlorophylls, pheophytins and quinones) and applied rigorous classical and quantum formalisms to calculate the energy surfaces for the primary ET in the bacteria Rhodobacter sphaeroides. In the latter investigation, the kinetic parameters for the electron transfer along the active (L) and inactive (M) branches have been determined. With no post-processing parameter fit, our modeling computes, for two different charge distributions, the driving forces for the transfer of an excess electron to the cofactors on the L-branch in good agreement with experiments. A multi-exponential kinetics of the primary charge separation is also predicted, consistent with experimentally observed kinetics. In a latest development we have investigated the kinetic of electron transfer between ferredoxin and photosystem I.
Fig. 3: Graphical representation of the reaction center of Rhodobacter sphaeroides. The amphiphilic detergent (LDAO) is represented by spheres, while stick and licorice representations are uses for water and the protein, respectively. ©CEA/M. Marchi
An extension of this research topic to larger membrane protein complexes such as photosystem II of the cyanobacterium Thermosynechococcus elongatus or type II light harvesting complex (LH2) of purple bacteria Rhodopseudomonas acidophila strain 10050 (Fig. 4) in different micellar environments are underway (coll. Dr. Robert Bruno, LBMS, CEA).

Fig. 4: Snapshots of the type 2 light harvesting complex from the purple non-sulfur bacteria Rhodopseudomonas acidophila strain 10050 embedded in a LDAO (a-b) or glycolipid micelles (c-d). Different peptides are in blue and red colors (blue and red) and pigments (bactériochlorophylles, green) and glycolipids (yellow). Water is not shown for visual clarity ©CEA/S.Abel.
Structure and dynamics of the hydration around proteins or surfactant assemblies
We are also interested in characterizing the effect of the environment on the properties of biological water around or inside active sites of proteins that plays a crucial role in their equilibrium structures, enzymatic functions, and phenomena such as molecular recognition and protein-proteins interactions. We have previously characterized the dynamics of biological water around globular proteins by extensive computer modeling. Our simulations have shown, for example, that the modern force fields and current molecular modeling approaches can reproduce the time scales of the dynamics of biological water as detected by the distribution of nuclear magnetic relaxation and other spectroscopic techniques. We were also able to elucidate the mechanism of attachment of water to the protein and the relationship between the rotational and translational diffusion of water. Our group has also developed models (implicit) to accelerate the sampling of the conformational space of a biopolymer and directly calculate the thermodynamic properties such as free energies. (coll. D. Borgis, ENS, Paris).
Our current researches are now devoted to a better understanding of the role of the environment or of confinement on the interfacial properties of water near different types of amphiphilic aggregates such as micelles, inverted micelles or biological membranes (coll. Dr. D. Laage, ENS, Paris and with Dr. F. Sterpone, IBPC, Paris, France).