The increasingly easy study of microbial ecosystems, known as microbiotes, offers new perspectives for applications in fields as varied as agriculture or human health, or in the fight against bioterrorism, the detection of emerging pathogens, and the characterization of their reservoirs.
Metaproteomics makes it possible to rapidly identify the protein content of a microbiota and to probe how it functions. Theoretically, it is also possible to establish the diversity of the microorganisms that make it up. The branches of life and taxonomic levels (strain, species, genus, family...) of peptides identified by mass spectrometry can be determined by comparing their sequences with those contained in molecular databases. But there are limits. The robustness and completeness of the database used directly influences the result, as most peptides are shared between microorganisms.
A team from the Laboratoire Innovations technologiques pour la Détection et le Diagnostic (LI2D, in Marcoule) of the Department of Medicines and Technologies for Health (DMTS) has just developed a mathematical method for identifying and measuring the organisms present in a microbiota. It consists in predicting for each living organism the number of peptides common to all the organisms present in the database and in considering that the whole signal obtained by mass spectrometry for a microbiota is in fact a combination of the signatures of all the organisms that make it up.
The researchers analyzed the proteome of the species Shigella flexneri by mass spectrometry (tandem MS/MS) and compared the peptide sequences to a database. They then determined the number of peptides in the database assigned to Shigella flexneri that they actually found in the spectrometry data, which they called the number of taxon-to-spectrum matches (TSM). They then made the same comparison again, considering not the base peptides assigned to Shigella flexneri but those assigned to other species, more or less phylogenetically distant from Shigella flexneri. Thus, a species very close to S. flexneri (such as Escherichia coli) has globally more peptides in common with it, and therefore a higher TSM number, than a species very distant (such as Bacillus subtilis). By comparing all the data, the researchers found a correlation between the number of TSMs and the phylogenetic distance between species. To validate the method and show that it is quantitative, the researchers then tested it by first analyzing artificial mixtures of two phylogenetically related organisms, S. flexneri and Salmonella bongori, with different proportions of the two, and then using more complex microbial models.
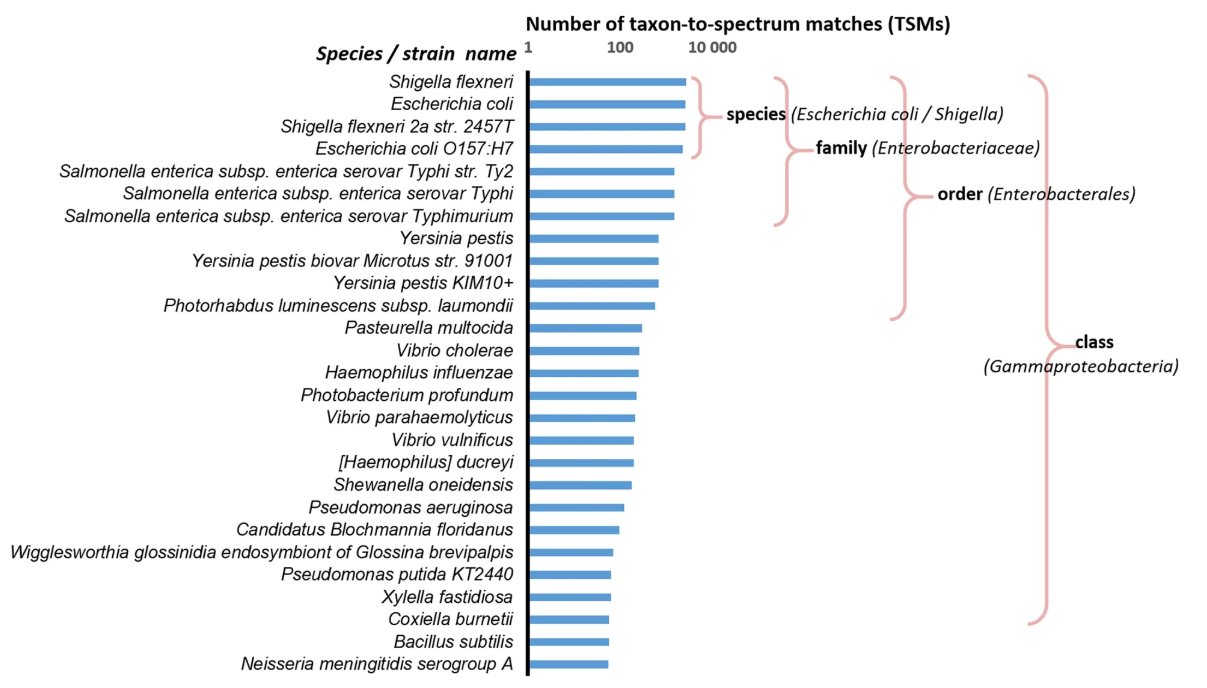
Number of taxon-to-spectrum matches for a pure Shigella flexneri sample. MS/MS spectra that can be associated with peptides and proteins to a selection of taxa available from a database (Ciccarelli et al.) were numbered. A key observation is that the number of taxon-to-spectrum matches attributed to taxa that are not in the sample is directly linked to the taxonomical proximity to Shigella flexneri
O. Pible et al. Microbiome, 2020
This new method of analysis, which they call phylopeptidomics, opens up new horizons for the study of microbiotes and their dynamics. The method can be applied to bacteria as well as to archaea and eukaryotes. It allows a quick and unbiased identification of any living organism, even in complex mixtures. Within the framework of the interministerial research and development program against the CBRN-E terrorist risk (Nuclear, Radiological, Biological, Chemical and Explosive), the laboratory is developing applications of this new method for the identification of all types of pathogenic agents without a priori.
Contact: Jean Armengaud (jean.armengaud@cea.fr)